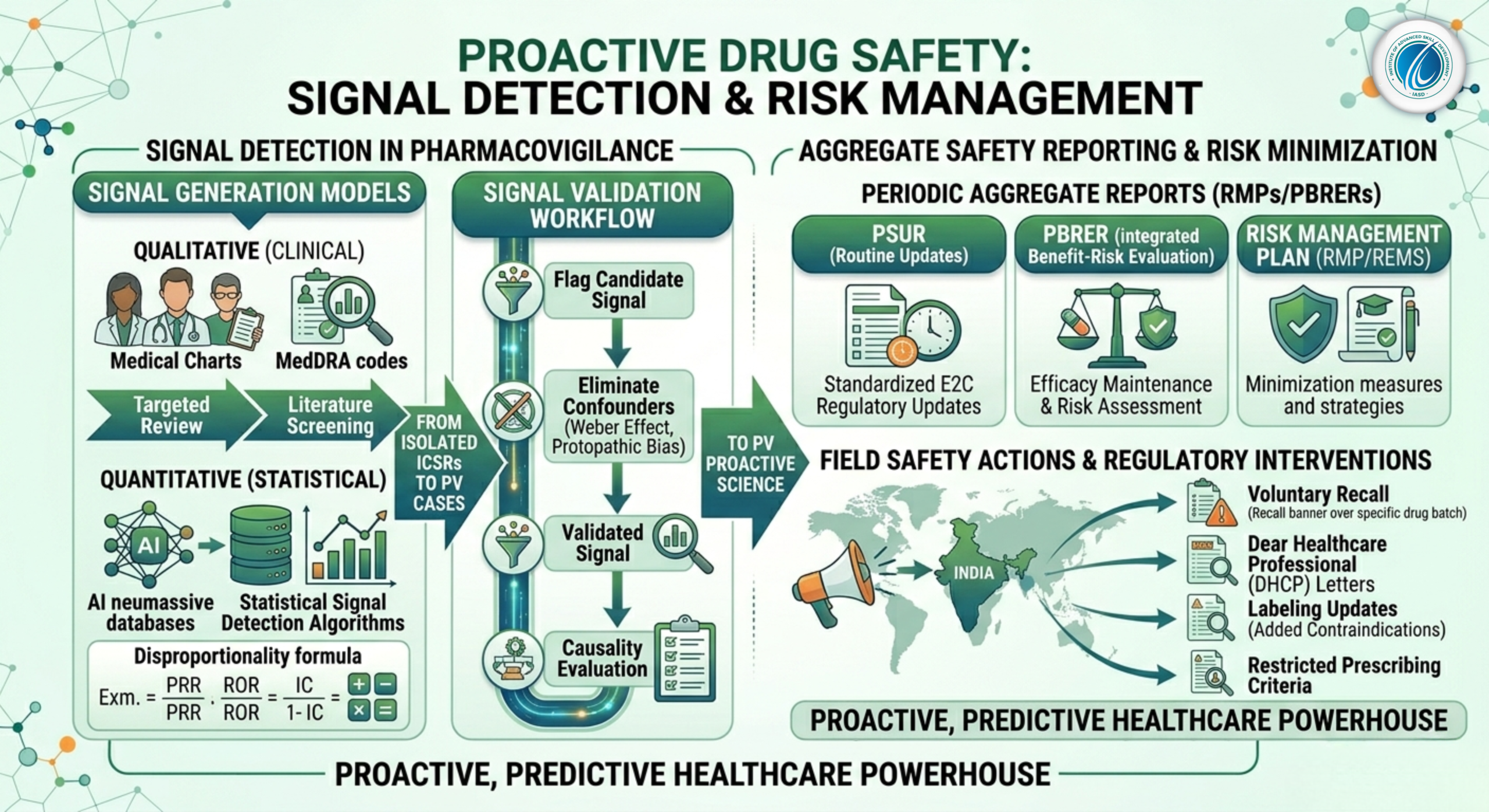
In the daily routine of drug safety processing, handling Individual Case Safety Reports (ICSRs) acts as the baseline defense system. Medical coding via MedDRA and narrative writing ensure that every individual adverse event is meticulously documented, validated, and archived. However, processing isolated cases is reactive by nature. If an organization only reviews individual reports, it might miss the broader patterns hiding within millions of data points.
The true strategic peak of drug safety monitoring is Signal Detection and Risk Management. This advanced discipline transforms pharmacovigilance from a reactive case-processing assembly line into a proactive, predictive science capable of identifying unknown risks, updating global prescribing laws, and executing life-saving public health interventions long before an outbreak or systemic crisis occurs.
1. Defining the Safety Signal: Beyond Isolated Cases
1.1 What Is a True Safety Signal?
According to the Council for International Organizations of Medical Sciences (CIOMS) and the European Medicines Agency (EMA), a Safety Signal is defined as reported information on a possible causal relationship between an adverse event and a drug, the relationship being unknown or incompletely documented previously.
A signal can manifest as:
-
A brand-new, never-before-seen adverse reaction associated with a therapeutic molecule.
-
An unexpected increase in the frequency, severity, or duration of a well-known, documented side effect.
-
The identification of a novel at-risk patient population (e.g., a reaction specific to pediatric patients, geriatric cohorts, or individuals with renal impairment).
1.2 The Lifecycle of a Safety Signal
A safety signal transitions through a strict, auditable multi-tier governance lifecycle before it results in regulatory action:
[Signal Generation / Detection] ➔ [Signal Validation] ➔ [Signal Analysis & Evaluation] ➔ [Recommendation for Action]
2. Signal Generation Methodologies: Quantitative vs. Qualitative
Modern safety databases capture unprecedented volumes of clinical information. To find true patterns within this data, pharmacovigilance departments combine qualitative medical insights with quantitative statistical algorithms.
2.1 Qualitative (Clinical) Signal Detection
Qualitative detection relies on expert clinical review, medical observation, and scientific intuition. It is highly effective for identifying strong causal relationships from small case series.
-
Targeted Medical Review: Drug safety physicians regularly screen clusters of serious unexpected cases, looking for distinct medical hallmarks (e.g., a specific pattern of acute liver injury occurring within 48 hours of drug administration across three unrelated patients).
-
Literature Screening: Systematically auditing international medical journals for published case reports or clinician-led independent safety reviews.
2.2 Quantitative (Statistical) Signal Detection
When dealing with massive global safety databases containing millions of records (such as the US FDA’s FAERS or the WHO’s VigiBase), manual human review across every drug-event combination is impossible. PV teams use statistical algorithms to scan databases for Disproportionality. These tools look for instances where a specific adverse event is reported significantly more often with a target drug than with all other medicines in the database.
Core Disproportionality Metrics
Pharmacovigilance statisticians use specific math models to analyze data:
-
Proportional Reporting Ratio (PRR): Compares the proportion of a specific adverse event relative to all events for a target drug against the same proportion for all comparison drugs.
-
Reporting Odds Ratio (ROR): An odds-ratio calculation checking if the odds of a specific event being reported with the target drug are statistically higher than the background population odds.
-
Information Component (IC): A highly sophisticated Bayesian neural network algorithm utilized by the Uppsala Monitoring Centre (WHO-UMC) to reduce statistical noise and prevent false alarms caused by low case counts.
3. The Signal Validation and Evaluation Workflow
Just because a statistical algorithm flags a disproportionality spike does not mean the drug caused the event. The flag is merely a candidate signal that must enter the strict Signal Validation Phase.
3.1 Eliminating Confounding Variables
During validation, safety scientists review the aggregate data to eliminate common statistical artifacts and clinical confounders:
-
Weber Effect Analysis: Accounting for the well-documented epidemiological pattern where adverse event reporting peaks sharply during the first two years of a new drug’s launch and then artificially drops off.
-
Protopathic Bias / Confounding by Indication: Investigating whether the adverse event was actually an early symptom of the underlying disease the drug was prescribed to treat (e.g., a patient taking an anti-cancer drug who develops severe fatigue, which is an intrinsic symptom of advanced malignancy).
-
Concomitant Medication Analysis: Cross-referencing if the flagged reaction only occurs when patients take the target drug alongside a specific secondary medication, indicating a drug-drug interaction rather than a standalone toxicity.
3.2 Formal Causality Evaluation
If a candidate signal survives validation, it is elevated to a Validated Signal. It then undergoes a formal medical evaluation, combining laboratory toxicology findings, biological plausibility analysis, and structural de-challenge/re-challenge results across the entire patient cohort.
4. Aggregate Safety Reporting: PSURs and PBRERs
While individual case safety processing operates on a daily timeline, international laws mandate that pharmaceutical companies periodically step back and evaluate their product’s global risk-benefit balance over time.
4.1 Periodic Safety Update Reports (PSURs)
The PSUR is a comprehensive, structured regulatory document compiled at specific intervals (e.g., six-monthly, annually, or every three years post-launch) that reviews all safety data gathered worldwide during that period. It serves to update health authorities on any emerging safety trends.
4.2 Periodic Benefit-Risk Evaluation Reports (PBRERs)
The PBRER represents the modern evolution of the PSUR under ICH E2C (R2) guidelines. The focus of a PBRER shifts from simply listing adverse events to conducting an integrated analysis of both safety and efficacy.
Focus of a PBRER Document
-
Efficacy Maintenance: Reviewing if new real-world clinical data confirms or challenges the efficacy metrics established during phase III clinical trials.
-
Integrated Benefit-Risk Analysis: Explicitly evaluating whether the drug’s therapeutic benefits continue to outweigh any newly identified safety risks across all approved indications.
5. Risk Management Plans (RMPs) and Regulatory Interventions
If a validated signal is medically confirmed as a true, undisputed adverse reaction, it becomes an Identified Risk. The pharmaceutical sponsor must design and submit a comprehensive Risk Management Plan (RMP) or Risk Evaluation and Mitigation Strategy (REMS) to outline how they intend to protect public health.
5.1 Routine Risk Minimization Measures
The first line of risk management involves updating the drug’s official regulatory documentation:
-
Updating the Summary of Product Characteristics (SmPC) and Package Leaflet: Adding the new reaction to the list of known side effects, establishing explicit contraindications, or adding a black-box warning.
-
Altering Prescribing Criteria: Restricting the drug’s use to specific lines of therapy or prohibiting its use in specific high-risk patient populations.
5.2 Additional Risk Minimization Measures (Educational Programs)
When labeling changes are not enough to guarantee safety, specialized educational campaigns are rolled out:
-
Dear Healthcare Professional (DHCP) Letters: Directly mailing urgent circulars to physicians, oncologists, and pharmacists to alert them to the newly confirmed risk and provide early management advice.
-
Physician Prescribing Checklists: Enforcing protocols where a doctor must complete a physical checklist verifying that a patient has undergone specific diagnostic tests (e.g., a baseline echocardiogram) before a high-risk medication can be dispensed.
5.3 Urgent Safety Restrictions and Product Recalls
If a validated signal reveals an extreme, unmanageable risk—such as severe organ failure occurring at standard therapeutic doses without any clinical warning—regulatory bodies or the manufacturer will implement an Urgent Safety Restriction. This results in the voluntary or legally mandated withdrawal of the product from the global market, triggering a comprehensive product recall to remove all active batches from hospital and pharmacy shelves.