Every medical breakthrough—whether a novel gene therapy, a targeted oncology drug, or a newly developed vaccine—relies completely on the validity of its clinical trial data. If a trial generates billions of data points that are messy, incomplete, poorly tracked, or filled with entries that contradict logic, regulatory agencies like the US FDA or EMA will reject the submission out of hand.
In clinical research, data integrity is paramount. Clinical Data Management (CDM) is the engine that converts unstructured clinical observations into a clean, high-quality, and structurally sound dataset ready for statistical analysis and regulatory approval.
1. Introduction to Clinical Data Management
1.1 What is CDM?
Clinical Data Management is a highly structured process within clinical trials that ensures the collection, integration, cleaning, validation, and delivery of high-quality, reliable, and statistically auditable patient data.
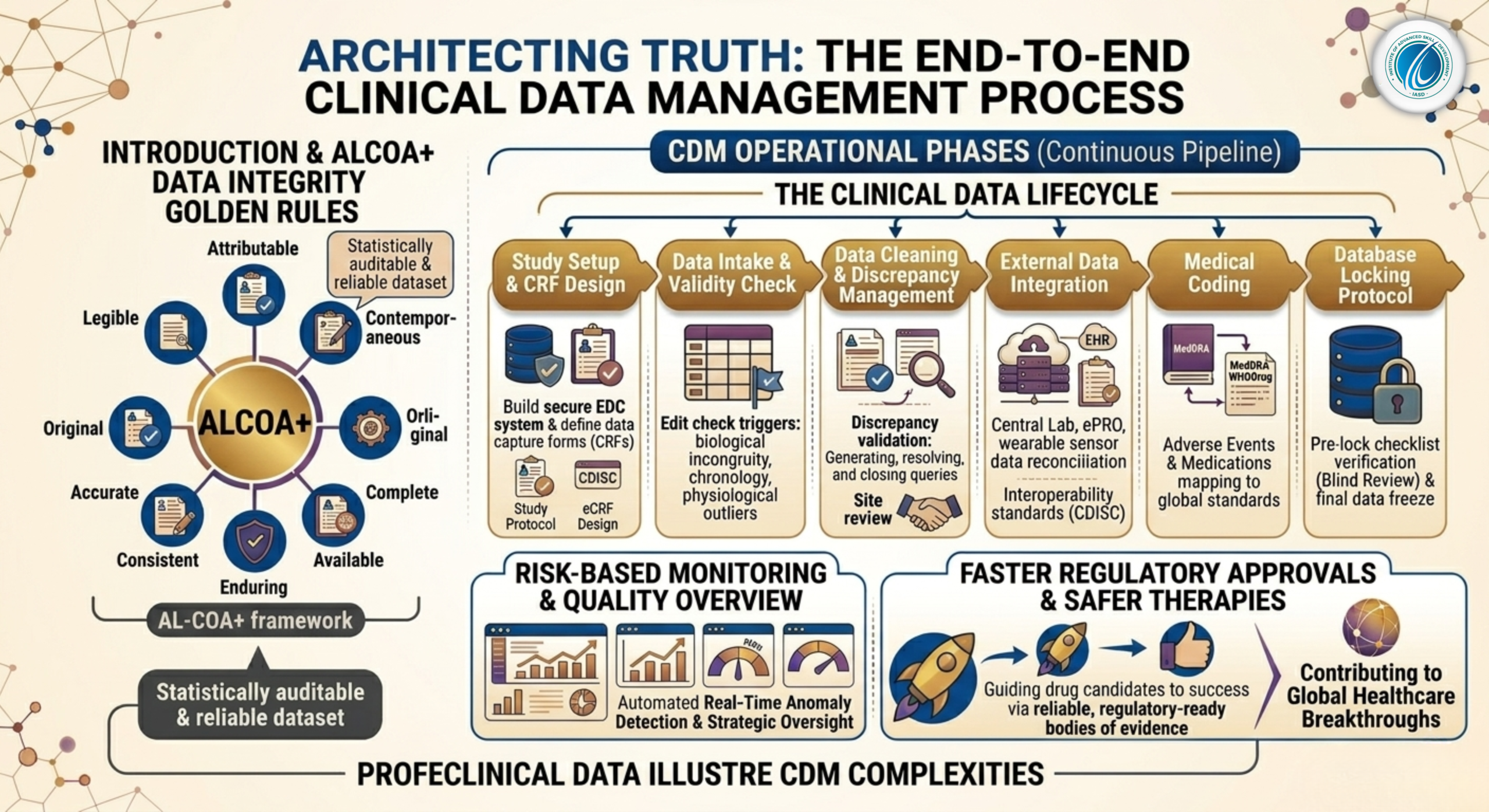
1.2 The Golden Rule: ALCOA+ Principles of Data Integrity
Every step of the clinical data lifecycle is governed by the international ALCOA+ framework, which defines the core standards for data integrity:
-
Attributable: Every data point must clearly show who recorded it, when it was entered, and from which source document it originated.
-
Legible: Data must be easy to read and understand across the entire life of the trial.
-
Contemporaneous: Data points must be logged at the exact time the clinical event or observation occurred.
-
Original: The data must be the first recorded instance of the information (or a certified true copy).
-
Accurate: Data must be completely free from entry errors, scientifically valid, and conform to reality.
-
Complete: No missing visits, unentered lab parameters, or unresolved data queries.
-
Consistent: The data must follow a logical, chronological sequence across all sections of the record.
-
Enduring: Records must be securely archived on durable media that remains accessible for decades.
-
Available: Data must be easily accessible for regulatory auditors or inspectors at any point during or after the study.
2. Phase 1: The Setup Phase (The Blueprint of the Study)
The clinical data management process begins long before the first patient is screened or randomized at a clinical site. The setup phase lays the structural foundation for how all data will be handled.
2.1 The Clinical Data Management Plan (DMP)
The Data Management Plan (DMP) is the ultimate regulatory blueprint for the trial’s data lifecycle. This extensive document defines every operational aspect of the study, including:
-
The exact electronic systems and software platforms to be used.
-
Detailed roles, responsibilities, and data access permissions across the study team.
-
Specific data transfer protocols for handling external datasets, such as central laboratory results or imaging files.
-
Comprehensive data storage, backup frequency, and long-term archiving strategies.
2.2 Case Report Form (CRF / eCRF) Design
The Case Report Form (CRF) is the specialized questionnaire used to collect all protocol-required data from each patient visit. Today, paper CRFs are largely obsolete, replaced by cloud-based Electronic Case Report Forms (eCRFs).
Data managers design eCRFs using standardized international libraries like CDISC CDASH (Clinical Data Interchange Standards Consortium – Clinical Data Acquisition Standards Harmonization). This ensures that data fields are defined identically across different trials globally.
2.3 Database Build and Edit Check Specifications
Once the eCRF design is finalized, database programmers build the clinical database inside an specialized Electronic Data Capture (EDC) platform (such as Medidata Rave, Oracle Clinical, or Veeva Vault CDMS).
A critical step here is drafting the Edit Check Specifications. Edit checks are automated validation scripts written into the database code to flag logical data entries in real-time.
Examples of Automated Data Validation Edit Checks
-
Biological Incongruity: Flagging an entry if a male patient is marked as pregnant, or if a prostate exam is logged for a female participant.
-
Chronological Discrepancies: Flagging an entry if a patient’s adverse event recovery date is logged as occurring before the drug was even administered.
-
Physiological Outliers: Flagging a heart rate entry of 650 beats per minute, or a body temperature of $22^\circ\text{C}$, prompting the site user to double-check their entry.
2.4 User Acceptance Testing (UAT)
Before a database goes live, it must undergo strict User Acceptance Testing (UAT). Data managers enter intentional errors, missing fields, and out-of-range dates into a test system to verify that every edit check triggers correctly and that the database architecture is secure.
3. Phase 2: The Conduct Phase (Data Cleaning and Query Management)
Once the trial begins, patient data streams in from hospitals and clinical sites worldwide. The conduct phase focuses on keeping this data stream clean, consistent, and validated.
[Data Entered into EDC] ➔ [Edit Check Triggers Flaw] ➔ [Data Manager Issues Query] ➔ [Site Investigator Resolves Query] ➔ [Data Manager Validates and Closes Query]
3.1 Data Review and Discrepancy Management
Despite automated edit checks, many complex errors require human review. Data managers run manual data listings to check for systemic inconsistencies across patient profiles. When a data anomaly is confirmed, it is flagged as a Discrepancy, and an electronic Query is sent through the EDC system to the hospital site.
3.2 The Query Resolution Lifecycle
-
Query Generation: The data manager issues a formal query explaining the issue (e.g., “Visit 3 lab values are missing. Please verify if the sample was drawn.”).
-
Site Review: The site’s clinical research coordinator or principal investigator reviews the query against the patient’s original medical charts.
-
Data Correction: The investigator updates the field in the EDC or adds an explicit explanation.
-
Query Closure: The data manager reviews the change against the study protocol. If the new entry satisfies logic, the query is closed, and the data point is validated.
3.3 External Data Integration and Reconciliation
Modern trials capture data from sources outside the direct EDC interface, including:
-
Central Laboratory Data: Electronic spreadsheets detailing chemistry, hematology, and biomarker panels.
-
eCOA / ePRO Platforms: Electronic Clinical Outcome Assessments and Patient-Reported Outcomes streamed directly from smartphones or patient tablets.
Data managers must run reconciliation scripts to ensure that dates, patient IDs, and sample collection times match perfectly between the external files and the internal EDC database.
3.4 Medical Coding via Standard Dictionaries
Unstructured medical events and medications entered by site coordinators must be coded into uniform terms using global dictionaries:
-
MedDRA: Used to standardize adverse events, medical history, and clinical diagnoses.
-
WHODrug Global: The premier dictionary used to map concomitant medications, over-the-counter drugs, and herbal supplements to their exact generic active ingredients and chemical classes.
4. Phase 3: The Closeout Phase (Securing the Final Dataset)
The closeout phase occurs when all patients have completed their treatment schedules and all study data has been successfully collected. This phase focuses on permanently freezing the data to prepare it for statistical analysis.
[All Queries Resolved] ➔ [Pre-Lock Checklist Verified] ➔ [Blind Review Completed] ➔ [Database Lock Approved] ➔ [Freeze Database Access]
4.1 Strict Pre-Lock Checklist Verification
Before a database can be locked, the clinical data management team must verify a comprehensive checklist:
-
All queries must be fully resolved and closed; no open discrepancies can remain in the EDC system.
-
All external laboratory and ePRO data must be fully integrated and reconciled.
-
All medical coding for adverse events and medications must be 100% complete and quality-checked.
-
All protocol deviations must be identified, categorized, and logged.
4.2 Data Blind Review
In double-blind clinical trials, where neither the patient nor the doctor knows who received the active drug or the placebo, a formal Blind Review is conducted just before database lock. The team evaluates the data while keeping treatment assignments hidden to ensure that data decisions are completely unbiased and objective.
4.3 The Database Locking Protocol
Once the pre-lock checklist is verified and signed off by the lead data manager, principal statistician, and medical monitor, the Database Lock is executed.
The database administrator revokes edit permissions for all users, freezing the entire dataset into a read-only state. This prevents any further modifications, ensuring that the final statistical analysis is performed on an unalterable, completely auditable, and regulatory-ready body of evidence.