The modern healthcare ecosystem relies heavily on an intricate network of biomedical engineering marvels. From simple diagnostic tools like tongue depressors and clinical thermometers to highly complex, life-sustaining innovations like automated insulin pumps, coronary stents, implantable defibrillators, and AI-driven hospital ventilator software, medical devices are foundational to patient care.
However, unlike pharmaceutical products that interact chemically with the human body’s metabolic pathways, medical devices present a completely different set of risk variables. A tablet cannot suffer from mechanical fatigue, software bit-rot, or a battery short-circuit. A medical device can.
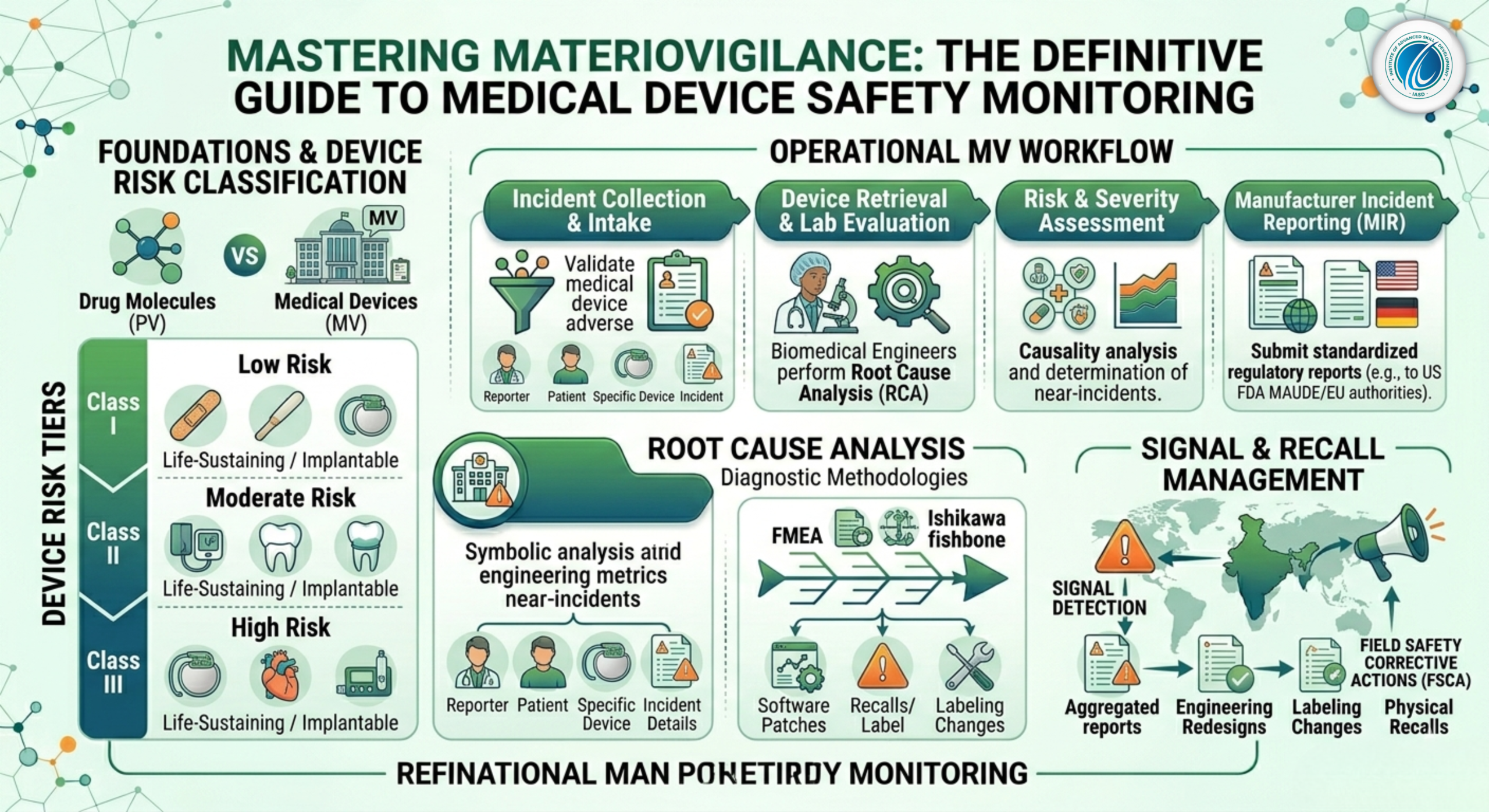
As a result, tracking and maintaining the safety of these technologies requires a highly specialized discipline distinct from traditional pharmacovigilance: Materiovigilance.
1. What is Materiovigilance? Definition and Core Concepts
1.1 Defining the Discipline
Materiovigilance is defined as the systematic surveillance, collection, evaluation, and reporting of adverse incidents related to the use of medical devices. The primary goal of this science is to protect patient health by preventing the recurrence of device-related mishaps across the global healthcare ecosystem.
1.2 Drugs vs. Devices: A Fundamental Risk Contrast
To understand why materiovigilance requires unique operational protocols, we must examine the differences between drug failures and device malfunctions:
| Feature Risk Profile | Pharmaceutical Drug Safety (PV) | Medical Device Safety (MV) |
| Primary Interaction | Systemic chemical, metabolic, or immunological mechanisms. | Physical, mechanical, electrical, or software execution. |
| Common Causes of Failure | Unpredicted toxicities, off-target drug interactions, idosyncratic metabolic mutations. | Mechanical wear, software bugs, material degradation, design flaws, user interface errors. |
| Root Cause Source | Often intrinsic to the patient’s individual biochemistry. | Intrinsic to the device design, manufacturing lot, or user training. |
| Corrective Action | Updating safety labels, adding contraindications, or drug withdrawal. | Software patches, hardware upgrades, engineering redesigns, or physical device recalls. |
2. Risk Classification Frameworks: Class I to Class III Devices
Regulatory frameworks do not treat all medical devices equally. Safety monitoring requirements scale exponentially based on the potential risk a device poses to a patient if it malfunctions. Under global standards like the US FDA and European Medical Device Regulation (EU MDR), devices are divided into three primary risk tiers:
2.1 Class I: Low Risk
These devices present minimal potential harm to users and are typically simple in design. They are subject to general regulatory controls but require less post-market tracking.
-
Examples: Manual toothbrushes, tongue depressors, surgical bandages, stethoscopes, and non-electric wheelchairs.
2.2 Class II (divided into IIa and IIb under EU MDR): Moderate Risk
These devices are more complex and require specific controls to ensure safety and efficacy. Malfunctions can result in non-life-threatening injuries or diagnostic errors.
-
Examples: Blood pressure monitors, contact lenses, surgical drills, ultrasound scanners, and dental crowns.
2.3 Class III: High Risk (Life-Sustaining or Implantable)
These highly sophisticated systems support or sustain human life, are implanted deep within bodily cavities, or present an unreasonable potential risk of illness or injury if they fail. Class III devices face strict post-market materiovigilance mandates.
-
Examples: Cardiac pacemakers, coronary stents, mechanical heart valves, automated external defibrillators (AEDs), and automated insulin delivery systems.
3. The Anatomy of a Medical Device Adverse Incident
At the operational core of materiovigilance is the processing of an Adverse Incident. Unlike drug processing, an incident report does not always require a patient to suffer an injury; a device defect that could have led to harm is enough to trigger a report.
3.1 The Three Criteria for a Reportable Incident
An event is considered a reportable materiovigilance incident if it satisfies three criteria defined by global competent authorities:
-
An event has occurred involving a medical device.
-
The medical device is suspected to be a contributory cause of the incident.
-
The event led, or might have led, to any of the following outcomes:
-
Death of a patient, user, or third party.
-
Temporary or permanent serious deterioration in a patient’s state of health.
-
A serious public health threat.
-
3.2 The Concept of a “Near-Incident”
A unique aspect of materiovigilance is the reporting of a Near-Incident. This occurs when a severe malfunction is detected by a healthcare professional before it can harm a patient. For example, if an intensive care ventilator displays a critical software freeze while being tested prior to patient connection, it is a reportable near-incident. The fact that no patient was hooked up does not eliminate the risk; the software flaw could manifest on an active patient in another hospital.
4. The End-to-End Materiovigilance Workflow
[Incident Collection] ➔ [Triage & UDI Tracking] ➔ [Root Cause Analysis] ➔ [Manufacturer Testing] ➔ [Regulatory Reporting] ➔ [Field Safety Actions (FSCA)]
4.1 Step 1: Incident Reporting and Intake
Incidents are flagged by hospital biomedical engineering units, clinicians, or patients directly to the device manufacturer’s safety division. Initial intake focuses on securing the device model, serial number, and lot number.
4.2 Step 2: Unique Device Identification (UDI) Tracking
Every modern medical device must carry a Unique Device Identification (UDI) barcode. The UDI consists of two parts:
-
Device Identifier (UDI-DI): A static code identifying the specific model and manufacturer.
-
Production Identifier (UDI-PI): A dynamic code tracking the specific lot number, batch number, manufacturing date, and software version.
During intake, specialists extract this UDI data to instantly cross-reference whether similar failures are occurring within the same manufacturing batch.
4.3 Step 3: Root Cause Analysis (RCA) and Laboratory Engineering Evaluation
Once an incident is logged, the physical device is retrieved from the clinic and returned to the manufacturer’s specialized testing laboratories. Biomedical engineers and safety analysts conduct a thorough Root Cause Analysis (RCA) using industrial methodologies:
Root Cause Analysis Diagnostic Methodologies
-
Failure Mode and Effects Analysis (FMEA): Proactively mapping out all potential sub-component failure points to calculate their downstream impact on patient safety.
-
The Ishikawa (Fishbone) Diagram: Systematically parsing whether a device failure stemmed from material raw defects, manufacturing calibration errors, software logic glitches, environmental variables (such as ambient heat), or user interface complexity.
4.4 Step 4: Medical Assessment and Severity Graduation
Simultaneously, medical directors evaluate the clinical impact of the failure. They determine if the injury resulted from a true engineering defect or a user error caused by poorly drafted Instructions for Use (IFU).
4.5 Step 5: Regulatory Submission
If the incident involves severe health deterioration or death, the manufacturer must compile a standardized Manufacturer Incident Report (MIR) and submit it electronically to relevant regional authorities (such as the US FDA’s Manufacturer and User Facility Device Experience [MAUDE] database or European competent authorities) within strict statutory timelines.
5. Field Safety Corrective Actions (FSCA) and Recalls
When root cause investigations confirm a widespread or high-risk systemic defect across a product line, manufacturers must execute a Field Safety Corrective Action (FSCA).
[Widespread Defect Identified] ➔ [Issue Field Safety Notice (FSN)] ➔ [Execute FSCA (Patch, Repair, or Physical Recall)]
5.1 Field Safety Notices (FSN)
An FSCA is accompanied by a Field Safety Notice (FSN), an urgent message sent directly to hospital directors, risk managers, and surgeons. The FSN outlines the defect, immediate mitigation steps, and how the manufacturer intends to fix the problem.
5.2 The Spectrum of Corrective Actions
An FSCA does not always mean physically pulling machines out of hospitals. Depending on the root cause, a corrective action can take several forms:
-
Software Updates: Deploying an over-the-air firmware patch to fix a software bug.
-
Component Retrofitting: Sending field engineers to replace a specific sub-assembly or battery lot across installed hospital systems.
-
Label Modifications: Updating the device’s physical labeling or user manuals to include new warnings or clear user instructions.
-
Physical Product Recalls: Permanently removing a device lot from the market due to an unfixable material defect or design flaw.
6. Global Regulatory Landscape: The Impact of EU MDR
The global materiovigilance framework changed dramatically with the full implementation of the European Medical Device Regulation (EU MDR 2017/745). This regulatory shift set a strict new global standard for device tracking.
6.1 Shift from Reactive to Proactive Monitoring
Historically, device safety relied on reactive reporting—waiting for a hospital to flag a failure. The EU MDR flipped this model, demanding aggressive, continuous Post-Market Surveillance (PMS). Manufacturers must actively gather real-world safety data through mandatory Post-Market Clinical Follow-up (PMCF) studies.
6.2 Periodic Safety Reporting (PSURs for Devices)
Under EU MDR, manufacturers of Class IIa, IIb, and Class III devices must periodically compile a formal Periodic Safety Update Report (PSUR). This report details all post-market surveillance data, safety signals, trend analyses, and FSCAs executed over a reporting period, ensuring accountability throughout the product’s market life.