The journey of a life-saving therapeutic molecule from a laboratory bench to a patient’s bedside is an incredibly complex, heavily regulated process that spans over a decade and costs billions of dollars. In traditional academic settings, students are taught that the approval of a drug by regulatory bodies such as the United States Food and Drug Administration (US FDA) or the European Medicines Agency (EMA) represents the ultimate finish line. However, within the industrial matrix of clinical research, that approval is merely the starting line of an entirely new, high-stakes discipline: Pharmacovigilance (PV).
While phase I, II, and III clinical trials are meticulously designed to evaluate a drug’s efficacy and safety, they possess inherent structural limitations. Clinical trials are conducted on a highly selected, homogeneous cohort of a few hundred to a few thousand patients. These individuals are monitored in controlled environments, free from common real-world variables such as severe comorbidities, complex concomitant medications, genetic diversities, and lifestyle variations.
The real test of a drug begins when it is unleashed onto the global commercial market and consumed by millions of diverse patients. This critical transition is exactly why pharmacovigilance exists.
1. What is Pharmacovigilance? Foundations and History
1.1 The Definition and Scope of Drug Safety
According to the World Health Organization (WHO), Pharmacovigilance is defined as the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems. The scope of modern PV has expanded far beyond simple adverse event collection to include:
-
Substandard and Falsified Medicines: Identifying counterfeit or poorly manufactured products in the supply chain.
-
Medication Errors: Analyzing mistakes in prescribing, dispensing, or administering drugs.
-
Lack of Efficacy Reports: Investigating instances where a well-established medicine fails to produce the expected therapeutic effect.
-
Misuse and Abuse: Tracking off-label usage patterns and intentional drug abuse.
1.2 Historical Milestones: From Thalidomide to Modern Regulation
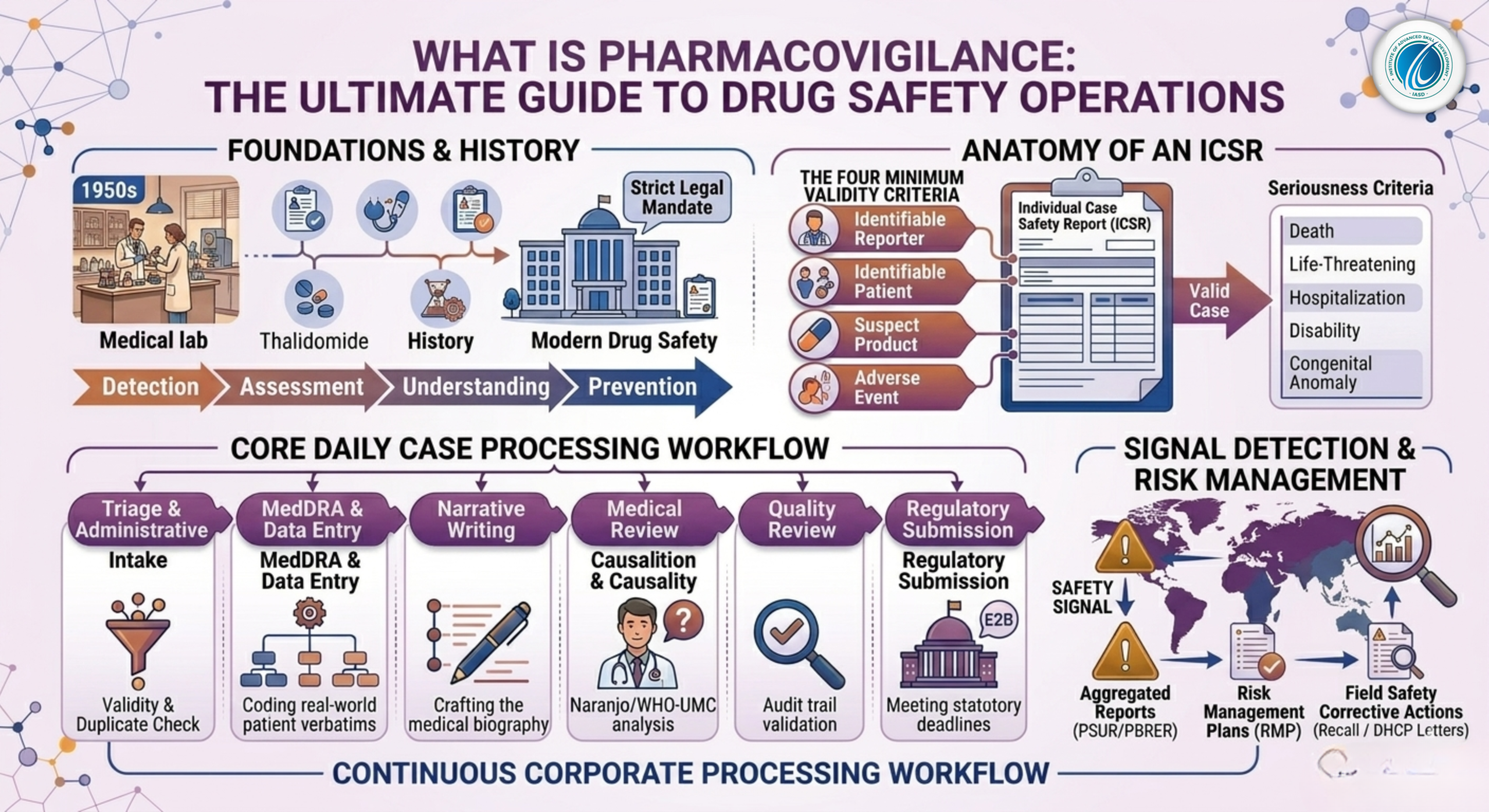
The contemporary structure of drug safety was built on lessons learned from historical public health crises. The most defining catalyst occurred in the late 1950s and early 1960s with the Thalidomide Disaster. Thalidomide, a sedative marketed to pregnant women to treat morning sickness, caused severe phocomelia (congenital limb deformities) in more than 10,000 children globally.
This tragedy completely altered the global regulatory landscape, forcing governments to establish systematic post-marketing surveillance networks. Today, pharmacovigilance is not a voluntary corporate practice; it is a strict legal mandate enforced by global health authorities to prevent similar public health catastrophes.
2. The Anatomy of an ICSR: Individual Case Safety Reports
The fundamental operational unit of daily pharmacovigilance operations is the Individual Case Safety Report (ICSR). An ICSR is a standardized document that details an adverse event experienced by an individual patient who was administered one or more specific medicinal products at a given time.
2.1 The Four Minimum Criteria for a Valid Case
Before a drug safety associate can process a safety complaint into a global database, the report must satisfy the Four Minimum Criteria for Case Validity. If even one of these elements is missing, the case is deemed invalid and cannot be transmitted to regulatory bodies:
-
An Identifiable Reporter: There must be a verifiable source who provided the information (e.g., a physician, pharmacist, nurse, patient, or family member). Contact details are necessary to facilitate follow-up inquiries.
-
An Identifiable Patient: The report must contain descriptive indicators establishing that a real individual experienced the event (e.g., patient initials, age, date of birth, or gender).
-
At Least One Suspect Product: The report must explicitly name the active pharmaceutical ingredient (API) or brand name of the medicine suspected of causing the reaction.
-
At Least One Adverse Event: The patient must have experienced an untoward medical occurrence, sign, symptom, or abnormal laboratory finding.
2.2 Serious vs. Non-Serious Adverse Events
Once a case is validated, PV professionals must determine its Seriousness Criteria. This distinction dictates the regulatory reporting timelines. An adverse event is classified as Serious if it meets any of the following regulatory thresholds:
-
Results in patient death.
-
Is immediately life-threatening.
-
Requires initial hospitalization or prolongs an existing hospitalization.
-
Results in significant, persistent, or permanent disability or incapacity.
-
Causes a congenital anomaly or birth defect.
-
Constitutes an unfavorable medically important event based on clinical judgment (e.g., blood dyscrasias, seizures, or intensive care intervention).
Regulatory Timeline Note: Under International Council for Harmonisation (ICH) guidelines, all valid Serious Unexpected Adverse Reactions (SUSARs) must be fully processed, validated, and reported to health authorities within 15 calendar days (or 7 calendar days for fatal/life-threatening events). Non-serious cases typically follow a standard 90-day reporting cycle.
3. The Core Pillars of Daily Case Processing
Inside a corporate drug safety department, clinical data goes through a highly regulated, assembly-line process known as Workflow Case Processing.
[Raw Case Intake] ➔ [Triage & Validity Check] ➔ [Data Entry & MedDRA Coding] ➔ [Narrative Writing] ➔ [Medical Review] ➔ [Regulatory Submission]
3.1 Step 1: Triage and Validity Assessment
Triage is the gatekeeping stage of pharmacovigilance. During triage, senior safety associates review incoming faxes, emails, medical literature, and call-center logs. The primary goals are to establish case validity, determine seriousness, calculate the regulatory submission deadline, and eliminate duplicate reports.
3.2 Step 2: Comprehensive Data Entry and Administrative Capture
Once cleared by triage, the case enters the data entry phase. The associate manually or semi-automatically populates specific fields within a validated drug safety database (such as Oracle Argus Safety or ArisGlobal LifeSphere). This involves entering comprehensive demographic details, medical history, baseline laboratory diagnostics, treatment indications, exact dosage frequencies, and batch/lot numbers.
3.3 Step 3: Medical Coding via MedDRA
Patients and healthcare professionals describe medical issues in unstructured, highly variable everyday language. For instance, three different patients might describe their condition as “my head is pounding,” “severe throbbing hemicrania,” or “blinding headache.” To perform statistical analysis on millions of global cases, these descriptions must be converted into a uniform language.
This standardization is achieved using MedDRA (Medical Dictionary for Regulatory Activities). MedDRA organizes medical terms into a strict five-level hierarchy:
$$ \text{System Organ Class (SOC)} \rightarrow \text{High Level Group Term (HLGT)} \rightarrow \text{High Level Term (HLT)} \rightarrow \text{Preferred Term (PT)} \rightarrow \text{Lowest Level Term (LLT)} $$
A Practical Medical Coding Comparison Table
| Patient’s Raw Verbatim Description | MedDRA Preferred Term (PT) | MedDRA System Organ Class (SOC) |
| “Turned completely yellow and eyes are pale” | Jaundice | Hepatobiliary disorders |
| “Pins and needles feeling in left fingertips” | Paraesthesia | Nervous system disorders |
| “Heart feels like it’s racing and thumping” | Palpitations | Cardiac disorders |
3.4 Step 4: Medical Narrative Writing
The Safety Case Narrative is a complete, chronological medical biography of the patient’s experience. It synthesizes complex, fragmented clinical data points into a cohesive story. A high-quality narrative must be objective, logical, and structured to allow a regulatory reviewer to evaluate the case without looking at the raw database fields.
A Professional Case Narrative Framework
-
The Lead Sentence: Summarizes patient demographics, validation criteria, and the core reaction (e.g., “This is a valid, medically confirmed, serious spontaneous case report concerning a 54-year-old male patient who developed acute myocardial infarction while receiving Drug X.”).
-
Medical History and Baseline: Details underlying diseases, risk factors, allergies, and concurrent medications.
-
Suspect Product Administration: Outlines exact dates, strengths, batch numbers, indications, and therapy durations.
-
The Adverse Event Timeline: Chronologically details the onset of signs, diagnostic interventions, and lab values.
-
De-challenge and Re-challenge Results: Documents whether the symptoms stopped when the drug was discontinued (Positive De-challenge) or if they returned upon reintroduction (Positive Re-challenge).
-
Concomitant Therapy Details: Evaluates other drugs that could potentially explain the event.
-
Current Outcome: Concludes with the current status of the patient (e.g., recovered, recovering, fatal, or unknown).
3.5 Step 5: Medical Review and Causality Assessment
The finalized case is sent to a Drug Safety Physician (DSP) for official medical review. The physician conducts a deep clinical assessment to determine Causality—evaluating whether there is a reasonable baseline probability that the suspect drug caused the adverse event, or if it was driven by co-existing diseases or concurrent therapies. Physicians commonly utilize standardized evaluation algorithms like the Naranjo Adverse Drug Reaction Probability Scale or the WHO-UMC Causality Assessment Criteria.
4. Signal Detection and Risk Management
Processing individual cases is crucial, but the ultimate strategic objective of pharmacovigilance is macro-level data analysis: Signal Detection.
4.1 What Constitutes a Safety Signal?
A Safety Signal is defined as reported information on a possible causal relationship between an adverse event and a drug, the relationship being unknown or incompletely documented previously.
PV teams use statistical algorithms (disproportionality analyses like Proportional Reporting Ratios [PRR] and Reporting Odds Ratios [ROR]) to screen data for unexpected spikes in specific drug-event combinations.
4.2 Aggregated Safety Reports (PSURs and PBRERs)
In addition to processing individual cases, pharmaceutical companies must periodically compile aggregated safety data. These extensive documents evaluate the global risk-benefit balance of a therapeutic product over time:
-
Periodic Safety Update Reports (PSUR): Standardized safety summaries compiled at regular intervals for regulatory review.
-
Periodic Benefit-Risk Evaluation Reports (PBRER): Comprehensive, data-driven documents evaluating whether a drug’s therapeutic benefits continue to outweigh its newly identified safety risks.
4.3 Risk Management Plans (RMP)
If a safety signal is confirmed as a true risk, pharmacovigilance experts draft a Risk Management Plan (RMP) or a Risk Evaluation and Mitigation Strategy (REMS). These plans outline the actions required to minimize patient exposure to danger, which may include:
-
Adding explicit black-box warnings, contraindications, or side effects to the Product Package Insert.
-
Issuing Dear Healthcare Professional Letters (DHCP) to directly alert doctors to the new risk.
-
Enforcing restricted distribution channels or requiring mandatory laboratory testing prior to drug prescription.
-
Voluntarily or legally executing a Global Product Recall if the safety risks outweigh any therapeutic value.
5. Navigating Your Pharmacovigilance Career Path in India
Driven by global outsourcing models, India has become the premier engine for international drug safety operations, making it an ideal destination for life science graduates.
5.1 The Corporate Career Ladder
A career in pharmacovigilance provides a highly structured, vertical corporate path within multi-billion dollar Contract Research Organizations (CROs) and multinational pharmaceutical companies:
[Drug Safety Associate Trainee] ➔ [Drug Safety Associate (DSA) I & II] ➔ [Senior DSA / Quality Reviewer] ➔ [Team Leader / Workflow Coordinator] ➔ [PV Manager / Director] ➔ [Global Safety Head]
5.2 How to Stand Out in the Entry-Level Job Market
Because competition for fresher positions at top-tier companies is fierce, graduates need more than just a basic life science degree to stand out. Employers look for foundational, day-one industry competencies:
-
Practical Knowledge of Database Architecture: Understanding the user interface, fields, and routing logic of platforms like Oracle Argus Safety.
-
Fluency in MedDRA Coding: Demonstrating an understanding of the 5-level hierarchical mapping logic.
-
Strong Clinical Narrative Skills: The ability to convert chaotic medical chart notes into structured, grammatically precise narratives.
By bridging the gap between university theory and corporate execution, aspiring drug safety professionals can secure high-paying, stable careers that contribute directly to global patient safety.